In this class we cover three topics:
1. Computational methods for modeling protein structures and interactions. We will cover tools and apps in the Rosetta protein modeling suite. https://www.ncbi.nlm.nih.gov/pubmed/18410248
2. Application of biosensors for high-throughput metabolic engineering. https://www.ncbi.nlm.nih.gov/pubmed/20367033
Using protein design, we can build biosensors which are basically genetically and computationally designed receptors that bind to specific target molecules. For a good discussion on olfactory receptors and protein design, you should check out the thesis research of Thras!
3. FoldIt, protein folding game. Hands on demo of the FoldIt video game. https://www.youtube.com/watch?v=lGYJyur4FUA
//////////////////////////////////////////////////////////////
COMPUTATIONAL HOMEWORK
The objective of the homework is to run the Rosetta protein structure prediction simulations and analyze the results. The idea is to get an understanding of how computational protein modeling works, looking at protein structures using a viewer (PyMol or Chimera or Rasmol) and making sense of the squiggles and wiggles. You'll also hopefully appreciate the computing power needed for biomolecular simulations.
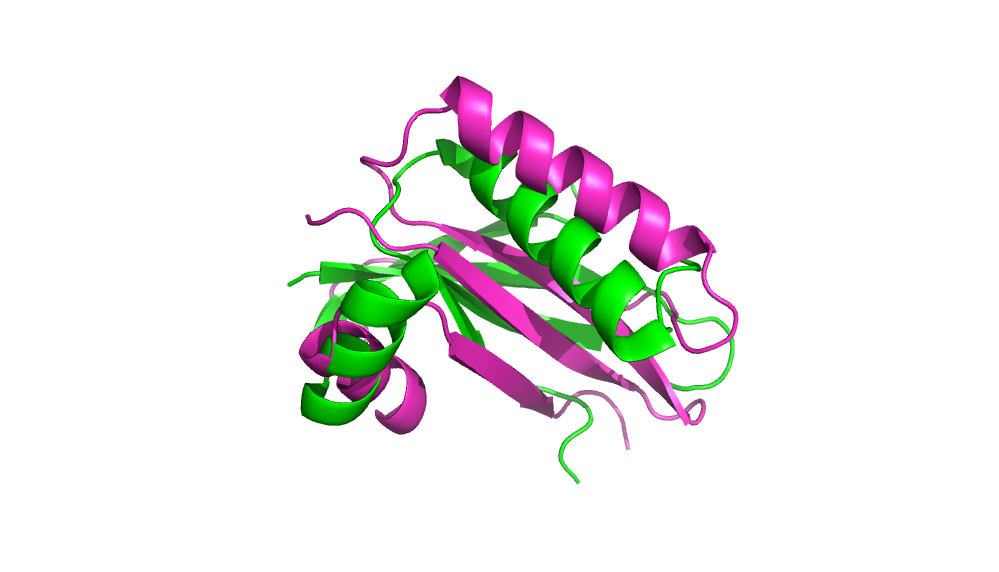
1. Pick a protein to perform ab initio folding. We picked 2HFQ.
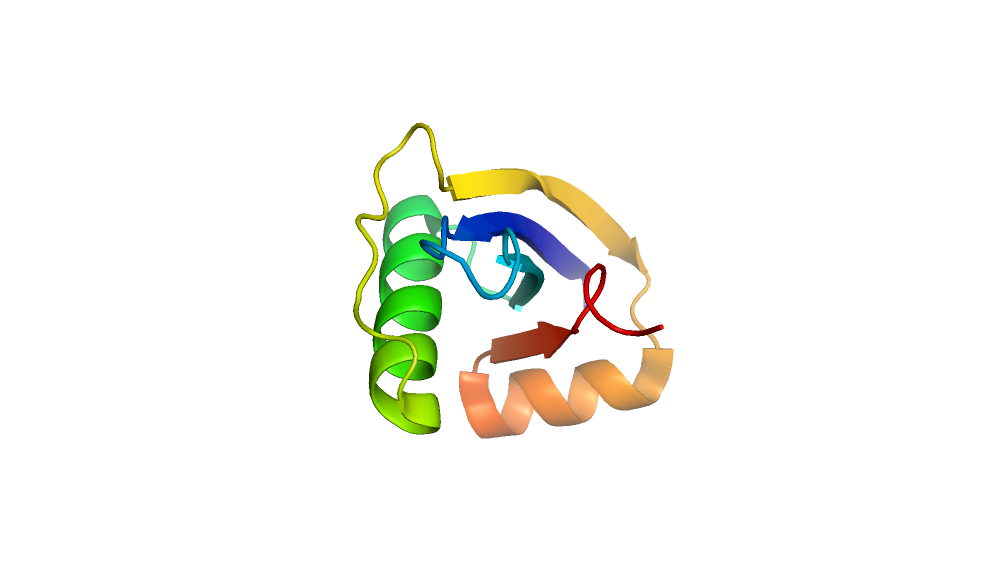
2. Generate models using AbInitio folding in Rosetta.
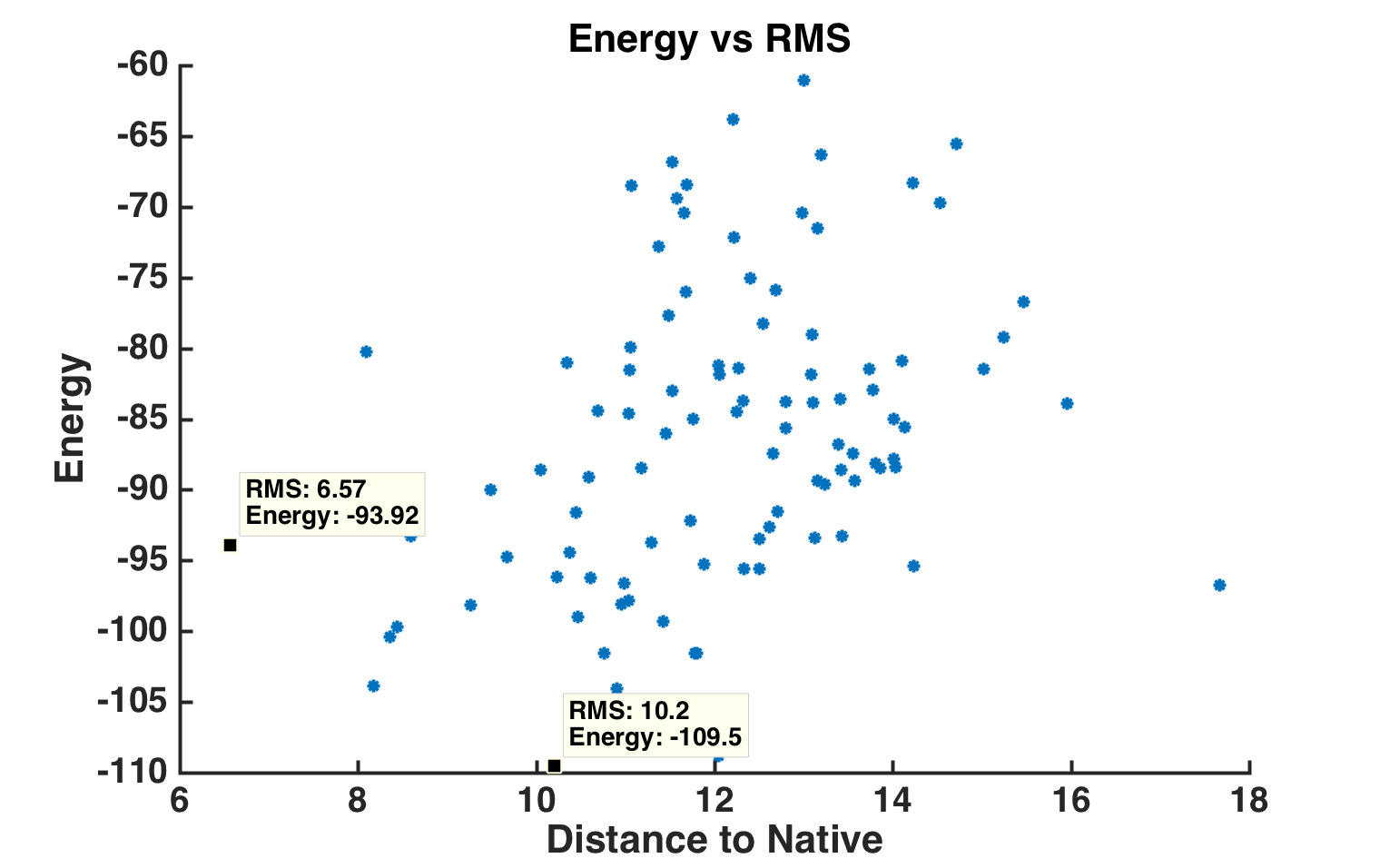
Energy vs RMS plot of 100 generated models.
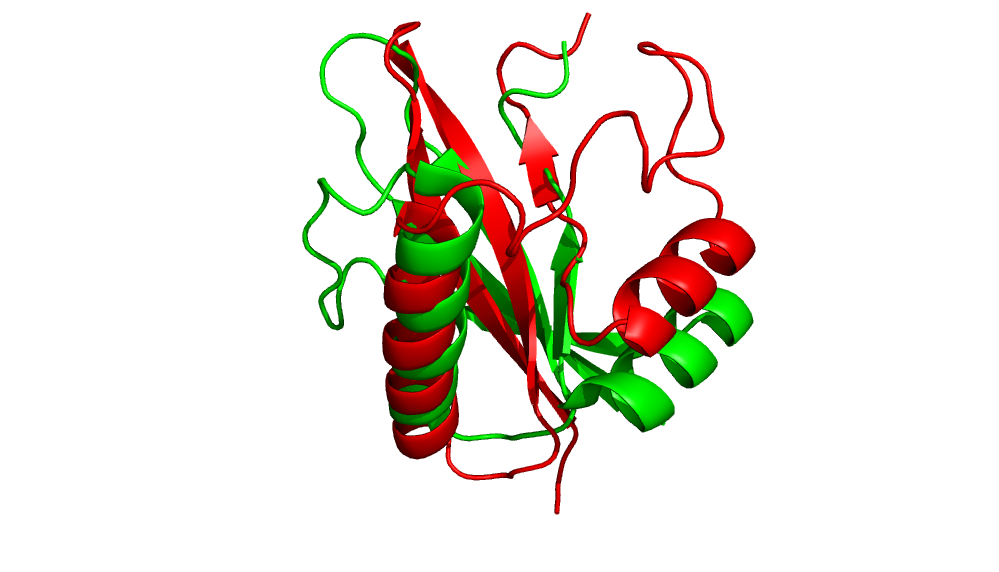
Comparison of minimum energy structure to native.
Comparison of minimum RMS structure to native.